-
Call Now
1800-102-2727

Process of Recombinant DNA Technology (Upstream processing), Practice problems and FAQs

What are the things that we consider while buying something in bulk? We definitely look for the places which can supply the bulk quantity of the products while maintaining product quality. We also look for the options where the product will be easily available.
How can a product of biological origin be created in bulk by considering all the above factors? There should be some technique to produce a large quantity of biological products such as enzymes, hormones, antibiotics, etc which have great potential for use in medical or commercial fields. The technique should be efficient enough to support the production even when demands for the product keep rising with time.
Insulin is such a product which is needed in a bulk quantity all over the world. But producing insulin in a lab is not a solution for this. Hence scientists found another option for this, which is the rDNA technology in which they manipulate the ability of bacteria to divide more in a single minute and also their ability to take up the foreign DNA into their plasmids. Recombinant DNA technology helps us to introduce foreign DNA containing the gene of interest which code for the specific products into organisms such as bacteria which multiply rapidly. This enables us to get the biosynthetic products in more amounts. But there are a lot of tools and techniques that are used in this process. In this article we will try and understand more about recombinant DNA technology and the process of upstream processing..
Table of contents:
- Recombinant DNA technology
- Upstream processing
- Practice problems
- FAQs
Recombinant DNA Technology
Recombinant DNA Technology is the technology used for introducing DNA sequence from one organism into another. Insulin is an example of a protein produced in large scale through rDNA technology.
Demonstration of rDNA technology was first done by Stanley Cohen and Herbert Boyer in 1972. These scientists came up with an idea of combining DNA of two different organisms. Cohen and Boyer chose two different bacteria to perform this experiment. One of the bacteria was E.coli and the other was Salmonella typhimurium. They found out a technique of inserting the antibiotic resistance genes found in the plasmid of S. typhimurium into the plasmid of E. coli. They used restriction endonuclease enzymes (enzymes which cut within the DNA at specific palindromic sequences) to cut the DNA fragment from the plasmid of S. typhimurium which contained the antibiotic resistance genes. The same restriction enzyme was used to cut the plasmid of E. coli and the DNA fragment from S. typhimurium was inserted into the cut part of the E. coli plasmid using DNA ligase. Thus a recombinant DNA or rDNA was created in vitro.

Fig: Stanley Cohen and Herbert Boyer (left to right)
Recombinant DNAs can be cloned and used to produce desired proteins expressed by specific genes of interest in large quantities. The process of large scale production of desired proteins includes two stages - upstream processing and downstream processing. Upstream processing is the initial phase in the bioprocess, which includes everything from early cell separation and cultivation to cell banking and culture development until the desired number is obtained. Downstream processing is the process after upstream processing. Downstream processing is for the recovery and the purification of biosynthetic products. Here we are going to discuss more about upstream processing.
Upstream processing
We can divide upstream processing into three steps - seed stock, scaling up and bioreactor.
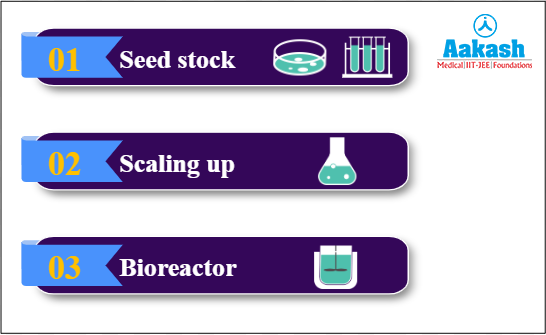
Fig: Steps of upstream processing
Seed stock is the complete procedure of forming the rDNA and getting the host ready and then inserting the GOI into the host. Scaling up is the process of preparing the stock for bioreactors. The final process is the preparation of the final product in bioreactors. Let us discuss each of these steps in detail.
Seed stock
In the process of getting the seed stock, there are many subprocesses. Those can be divided into: isolation of genetic material, cutting of DNA at specific locations, amplification of GOI, insertion of GOI in vector and insertion of rDNA into host.
Isolation of genetic material
First we have to find the gene responsible for production of our desired protein. This is a small portion of the entire genome. This is called the Gene of Interest (GoI), but in order to use it we must isolate the genomic DNA from the cell.
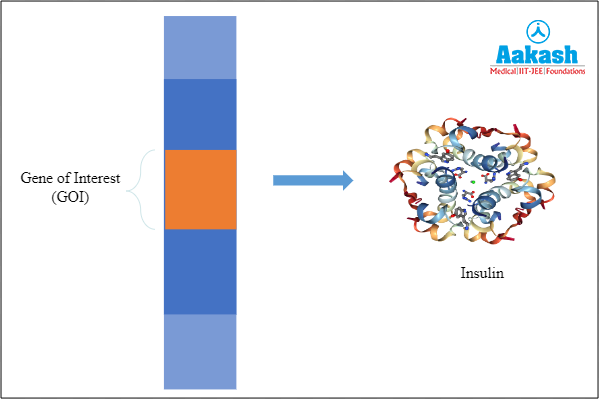
Fig: Gene of interest
To isolate the DNA from a cell, first the outer covering of the cell needs to be broken down. In the case of plant cells. fungal cells and bacterial cells, both cell wall and cell membrane need to be removed, using specific enzymes. In case of animal cells, only the cell membrane is present and is made of lipid bilayer. This can be easily removed using detergents.
There are specific enzymes used to break down the cell wall of different organisms based on the constituents of the cell wall. Lysozyme is used for the bacterial cells, cellulase for the plant cells and chitinase for the fungal cells.
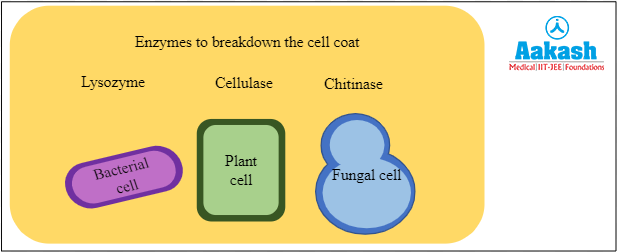
Fig: Different enzymes to break different cell coats
Detergents destroy the cell membrane and all lipids inside the cell, thus letting the cytoplasm leak. The cell has several other components like DNAand histones in the nucleus, debris from destroyed lipids, proteins and RNA in the cytoplasm. All these need to be removed as we need DNA in pure form.
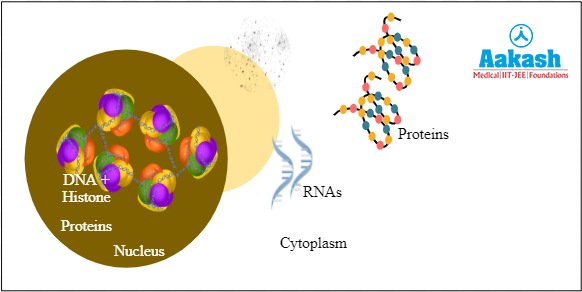
Fig: Components of cell
First we treat the cell with proteases that destroy the histone proteins and the proteins in the cytoplasm. Then we treat with Ribonucleases (Rnase) that destroy the RNA in the cytoplasm. But now there is still debris along with the DNA. To separate the DNA we need to add chilled ethanol to the cell homogenate. This will help the DNA to precipitate out of the mixture as a white thread like structure. A glass rod can be used to collect the DNA. This is known as spooling of DNA.
GIF: Addition of chilled ethanol

GIF: Spooling of DNA

Pure DNA can also be recovered by centrifuging the cell extract that is mixed with chilled ethanol. The pellet obtained has purified DNA. Now we have the DNA containing our gene of interest and the next step is to cut the genes in the isolated DNA solution.
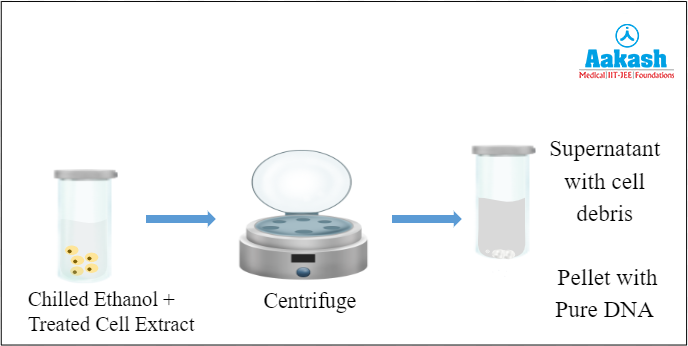
Fig: Centrifugation
Cutting of DNA at specific locations
Once the DNA is isolated, it is cut at specific locations using restriction endonuclease enzymes in order to cut out the GOI. To monitor the progress of restriction enzyme digestion, agarose gel electrophoresis is used. This technique involves passage of DNA through an agarose gel under the influence of an electric field. DNA being negatively charged, moves from the cathode (negatively charged terminal) to the anode (positively charged terminal). The pores of the agarose gel serve as a sieve to separate out the DNA fragments generated as a result of restriction endonuclease digestion. The smaller the fragment, the faster it moves towards the anode while the larger fragments lag behind.
A DNA ladder is also loaded onto the gel to serve as a reference point. It is a solution having DNA molecules of varying lengths that can be used as a reference to estimate the size of the separated DNA fragments. Once electrophoresis is over, the DNA fragments can be observed under UV light as orange bands. This is because the gel contains ethidium bromide which intercalates between the stacked bases in double-stranded DNA and fluoresces once exposed to UV light. Once a fragment having size equivalent to the GOI is observed it can be excised and eluted from the gel. This gives us our GOI.
GIF: Agarose gel electrophoresis

All of the above steps are also carried out to isolate the vector DNA. After obtaining the GOI we have to amplify it to obtain multiple copies. So the next step is the amplification of GOI.
Amplification of GOI
Kary Mullis solved this problem with the invention of a very special technique called PCR or Polymerase Chain Reaction. This helps to create many copies of the GOI, in vitro using two sets of primers and DNA polymerase enzymes.
When the process of DNA replication is done several times, a section of DNA can be amplified a billion times, resulting in 1 billion copies. The use of a thermostable DNA polymerase allows for repeated amplification. The three major steps of PCR are denaturation of the strands of the DNA template carried out at 94oC, annealing of primers to the 3’end of the template at 60oC, and extension of the primer to create complementary strands with the help of thermostable Taq polymerase enzyme at 72oC.
Thermocycler is the machine that allows multiple replication cycles of a particular fragment of interest from the whole genome.
Fig: Thermocycler

The amplified GOI has to be modified for the further steps. For that specific sequences which are identified by the restriction endonucleases are added to the amplified genes.
Insertion of GOI in vector
Now we have the modified GOI which can be inserted into the host organism. For that we need to insert the GOI into the vector. Both the vector and the gene of interest had been cut using the same restriction endonuclease enzymes and hence have complementary ends. The cut out 'gene of interest' from the source DNA and the cut vector with space are mixed and ligase is added to the mixture to join the two fragments. Recombinant DNA is created as a result of this process.
Fig: Formation of rDNA

Insertion of rDNA into the host organism
Bacteria are generally employed as hosts since they divide fast and create multiple copies of the gene of interest in a short time. Bacterial cell membranes are made up of hydrophobic phospholipids. DNA is hydrophilic. The charges of the DNA and the membrane phospholipids are also negative and they repel each other. Thus, the rDNA cannot pass through the bacterial cell membrane directly. Treating the cell with positive divalent ions like Ca2 + changes the charges on membrane and DNA, making it neutral.
Fig: Calcium rich environment helping the rDNA to

enter the bacteria
The cation rich environment makes the bacteria competent to take up DNA from its surroundings. Transformation is a procedure by which a foreign piece of DNA is taken up by a cell, usually bacterial, from the surrounding. Treating the cell with CaCl2 is a chemical transformation method. There are also other methods of transformation used to introduce the rDNA into the host cells like heat shock method (creating transient pores in the cell membrane by sudden heat shock), biolistics (using a gene gun to shoot microparticles of gold or tungsten coated with DNA into the cell nucleus), electroporation (creating transient pores in the cell membrane using electric shock), etc.
Once the process of transformation is complete, the transformed cells are selected using different techniques such as antibiotic resistance or blue white selection method. The selected cells can be used to prepare the seed stock.
Scaling up
In this step the culture from the seed stock is taken and it is then emptied into a flask containing fresh medium.
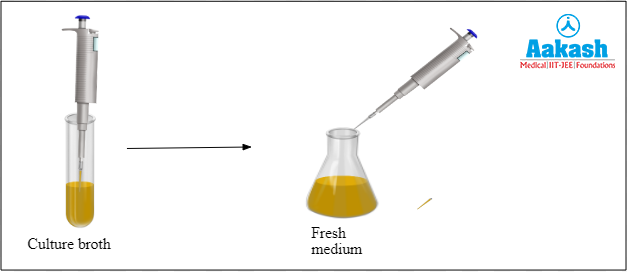
Fig: Transfer of cell culture into fresh growth medium
The flask with the culture is then kept in a shaker incubator where all the required optimal conditions are maintained. This produces a good amount of stock for culturing several litres of fresh medium.

Fig: Shaker incubator
Bioreactor
Bioreactors can be thought of as vessels (100-1000 litres) in which raw materials are biologically converted into specific products like proteins, individual enzymes, vitamins etc., using microbial, plant, animal or human cells. A bioreactor provides the optimal conditions for achieving the desired product by providing optimum growth conditions (temperature, pH, substrate, salts, vitamins, oxygen).
The stirred tank bioreactor is the most common type of bioreactor used in today’s time. The reactor tank is cylindrical and usually curved at the base to facilitate mixing. The stirrer facilitates mixing of the media. It also helps in mixing the oxygen available in the tank for better growth and avoids clumping of cells. Air is bubbled through the bioreactor using an aerator which passes sterile oxygen into the reactor. This is also called sparging. This maintains the optimum oxygen level in the reactor. There can be reactors without spargers. There is usually a foam control system to prevent foam formation due to constant stirring. Other than these features, it has a pH control system and temperature control system to maintain the optimum conditions for growth. There is also a sampling port which is used to withdraw small volumes of culture periodically to analyse the culture.
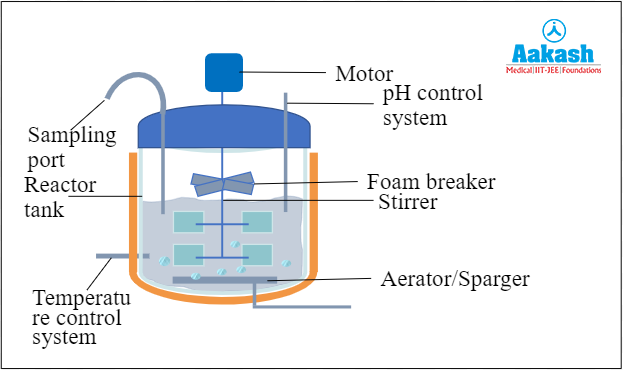
Fig: Stirred Tank Bioreactor
Bioreactors can be classified into two based on media supply. They are single batch bioreactor and fed batch or continuous bioreactor.
Single batch culture
Single batch culture method involves a closed system where all the nutrients and necessary conditions for growth are provided only once. Nothing is added or removed until the entire process is complete.
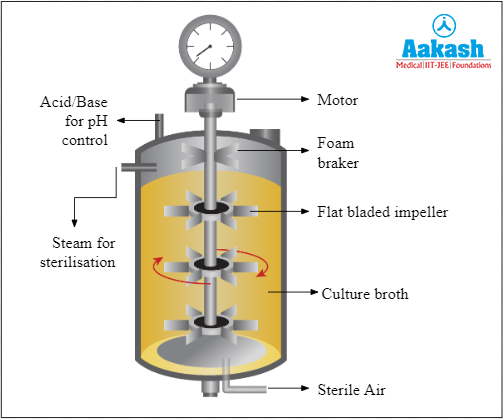
Fig: Single batch bioreactor
The organisms cultured in such a bioreactor follow all 3 phases of growth i.e an initial lag phase, log phase or phase of exponential growth and the stationary phase during which rate of cell growth and death becomes equal.
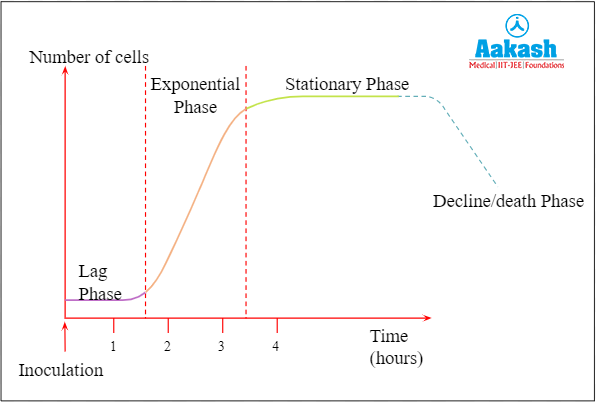
Fig: Phases of growth
Single batch cultures have little risk of contamination since everything is added at once. However it is costly and time consuming.
Fed batch culture
Fed batch culture method involves an open system where medium is added and culture is periodically removed from the reactor. Doing so maintains the microorganisms in their exponential growth phase. This process is cost effective and takes less time but the chances of contamination is very high.
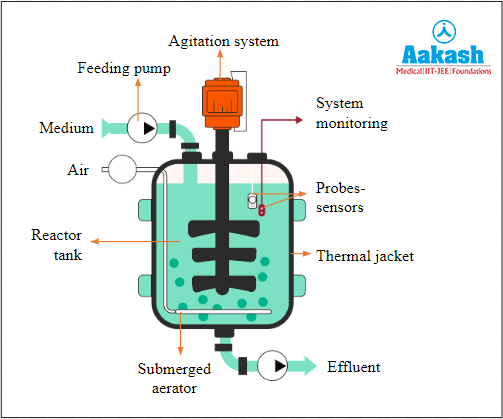
Fig: Fed batch bioreactor
Now that we have the biosynthetic product after the processing inside the bioreactor, it has to be recovered and purified from the growth medium. Hence it undergoes downstream processing. After this, the purified product is mixed with preservatives and undergoes clinical trials to get marketed for human use.
Practice Problems
Q 1. Various enzymatic treatments are used to break down the cell wall (if present), cell membrane, and other macromolecules in order to separate DNA from the cell. To dissolve the peptidoglycan layer in the cell wall, a cell is treated with a specific enzyme. This cell will be equipped with
a. membrane bound cell organelles
b. 80S ribosomes
c. Plasmids
d. nuclear membrane
Answer: Various enzymes are employed to dissolve the cell wall, cell membrane, and other macromolecules (proteins, lipids, RNA) in order to separate DNA from a cell. The cell employed to isolate DNA is a bacterial cell because peptidoglycan (murein) is found in the cell wall of bacteria. The peptidoglycan in the bacterial cell wall is normally degraded by the enzyme lysozyme. A bacterial cell is a type of prokaryotic cell that lacks membrane-bound organelles and a distinct nucleus (nuclear membrane is absent). In bacterial cells, 70S ribosomes can be found. Eukaryotic cells have 80S ribosomes in their cytoplasm. Plasmids are extrachromosomal circular DNA rings present in certain bacterial cells that replicate autonomously.
Hence, the correct option is c.
Q 2. Choose the option that correctly determines whether the provided assertions are true or false.
1. The thermocycler is the name of the instrument used in PCR.
2. The copies of DNA created by PCR are not identical, and each copy has minor differences.
3. PCR exponentially amplifies DNA strands.
a. I - T, II - F, III - T
b. I - F, II - T, III - F
c. I - T, II - T, III - F
d. I - T, II - F, III - F
Answer: PCR or polymerase chain reaction is a technique of amplifying a DNA sequence by creating millions of identical copies of the DNA sequence. For successful PCR amplification, the reaction mixture must be rapidly passed through various temperature levels. This is accomplished with the help of a thermocycler, which can quickly switch the reaction conditions between different temperatures at precise time intervals. In each PCR cycle, the number of DNA molecules doubles. 2n (n = number of PCR cycles) is the formula for calculating the number of DNA molecules generated after a certain number of cycles. For example, if we performed 32 rounds of PCR, the total number of double-stranded DNA molecules would be 232. The amount might be in the billions. As a result, it can be stated that PCR amplifies DNA at a rate that is exponential.
Hence the correct option is a.
Q 3. What is the primary purpose for using PCR in rDNA technology?
a. The amount of DNA extracted from a sample is usually relatively little.
b. It is sometimes impossible to extract DNA from cells.
c. PCR increases the chance of DNA isolation
d. PCR helps in generating fragments containing the gene of interest
Answer: Cell DNA extraction is sensitive, and there is a considerable risk of contamination. Following an appropriate technique, a variety of enzymatic treatments are used. Even then the amount of DNA extracted from a cell sample is usually insignificant. As a result, polymerase chain reaction (PCR) is utilised to increase the yield. This method generates replicas of the gene of interest using the enzyme DNA polymerase (which comes in thermostable versions). Primers that are targeted to specific DNA sequences of the template DNA are allowed to anneal to the denatured strands of the template. After that, the DNA polymerase extends the primers and creates complementary strands to create replicated daughter strands. This process is repeated in multiple cycles to obtain a large number of copies of the gene of interest.
Hence the correct option is a.
Q 4. The DNA containing the gene of interest from an animal cell must be clean and devoid of unwanted macromolecules in order to be isolated. DNA molecules are entangled with histone proteins. Which of the enzyme treatments will aid in the purification of DNA from its associated proteins?
a. RNase
b. Protease
c. Chitinase
d. DNase
Answer: The technique of extracting purified DNA from a cell is known as DNA extraction. It entails a number of processes involving enzymatic treatments. The removal of the cell wall is not required in an animal cell. The cell membrane is disrupted to cause cell lysis. This causes the lysis buffer to liberate all of the cellular components. To digest and eliminate any traces of RNA, RNase treatment is used. Nucleosomes are structures in which DNA is wrapped around proteins. It is necessary to treat DNA with a protease enzyme in order to obtain protein-free DNA. DNase treatment is not used during the DNA extraction process since it will digest the DNA.
Hence the correct option is b.
FAQs
Q 1. Who is credited as the inventor of genetic engineering?
Answer: The "Father of Genetic Engineering," Paul Berg, is well-known. Paul Berg pioneered genetic engineering in 1972. He was able to transfer an SV-40 virus gene into a bacterium with the help of the lambda phage.
Q 2. What are the differences between plasmid and vector?
Answer: The fundamental distinction between plasmid and vectors is that the former is an extrachromosomal element found mostly in bacterial cells, whilst the latter is a vehicle that transports foreign DNA molecules into another cell. Plasmids can be employed as vectors as well.
Q 3. What was the first rDNA-based product?
Answer: The first product of rDNA technology is humulin. Humulin, Eli Lilly's recombinant insulin manufactured from Genentech's highly engineered bacteria, was approved by the Food and Drug Administration in 1982. It was the first recombinant DNA medicine, as well as one of the first genetically altered products offered to the general public.
Q 4. What is the role of recombinant DNA in agricultural research?
Answer: Recombinant DNA has boosted plant growth in agriculture by enhancing nitrogen fixation efficiencies and copying and inserting bacterial genes into plant cells. Plants have been made immune to caterpillars, pests, and viruses by inserting resistance genes into their genomes.
YOUTUBE LINK: https://youtu.be/qSeuqV_lDwY
Related Topics
|
Principles of biotechnology, Practice Problems and FAQs |
|
Process of recombinant DNA technology: (Downstream processing), Practice problems and FAQs |
|
Transformation, Practice problems and FAQs |
|
PCR |
|
Cloning vectors, Practice Problems and FAQs |
|
Gel electrophoresis, Practice Problems and FAQs |
|
Restriction enzymes, Practice problems and FAQs |