-
Call Now
1800-102-2727


Mendelian Disorder: Haemophilia, Colour Blindness, Sickle Cell Anaemia, Thalassemia, Phenylketonuria, Cystic Fibrosis, Practice problems and FAQs
Have you noticed that whenever we get injury, bleeding occurs from the site of injury and then it automatically stops after a while. Do you know why? This is because the platelets and clotting factors in our blood help in coagulation of blood at the site of injury to prevent any major blood loss.
Now, think what happens to those people whose blood does not coagulate on its own?
Such people can have a lot of blood loss even from a minor cut and this condition is known as haemophilia. It is a type of genetic disorder that occurs due to mutation in a gene that codes for a blood clotting factor.
Similarly, there are other genetic disorders in humans that occur due to mutation in genes present on the autosomes or sex chromosomes. These are known as Mendelian disorders and are inherited by the offspring from their parents during sexual reproduction. In this article we will discuss some of the more commonly occurring Mendelian disorders and the genetic basis of their occurrence.
Table of contents
- Genetic disorders
- Mendelian disorders
- Haemophilia
- Colour blindness
- Sickle cell anaemia
- Thalassemia
- Phenylketonuria
- Cystic fibrosis (in brief)
- Practice Problems
- FAQ’s
Genetic disorders
Genetic disorders are defects that are caused due to defects in the genetic machinery of an individual such as genetic mutations, chromosomal anomalies and chromosomal aberrations. These disorders are inheritable from one generation to the next. The genetic disorders are broadly classified into two groups:
- Mendelian disorders - caused due to mutations in a gene which can result in the expression of altered proteins or no expression of certain proteins. This can in turn reflect in the form of major disorders in the body.
- Chromosomal disorders - caused due to absence or excess of chromosomes or abnormal arrangement of chromosomes in the cells of an organism. These disorders do not follow mendelian patterns of inheritance and hence cannot be predicted using pedigree analysis. A person with chromosomal disorders may or may not transmit the disorder to his or her offspring.
Mendelian disorders
Mendelian disorders occur mainly by the alteration or mutation in a single gene. The inheritance of these disorders follows the principles of inheritance as described by Mendel. Pedigree analysis can be used to trace the pattern of inheritance of Mendelian disorders in a family. Mendelian disorders are of two types:
- Sex-linked disorders
- Autosomal disorders
Sex-linked disorders
Sex-linked disorders are those which are caused by mutations in genes that are linked with sex chromosomes i.e. either with X-chromosome or Y-chromosome. Any defective gene that is inherited via the sex chromosomes are considered to be sex-linked. Sex-linked disorders are either dominant or recessive. In dominant inheritance, the defective gene received from one of the parents is dominant and is capable of causing disease in the offspring even in the presence of a normal allele in the homologous pair that is received from the other parent. On the contrary, for a sex-linked recessive disorder, the defective gene is recessive and is capable of expressing the disease in a person only if he or she inherits two recessive defective alleles, one from each parent. Examples of such disorders would include haemophilia, colour blindness, etc.
Autosomal disorders
Autosomal disorders are a type of disorders that are linked with genes present on autosomes or chromosomes other than sex chromosomes. They are not sex specific. These disorders can affect children of any sex. These disorders are either dominant or recessive. In dominant autosomal disorders, the defective allele is dominant and expresses the disease even in the presence of a recessive normal allele. In recessive autosomal disorder, the defective gene is recessive and can express the disease only in homozygous condition, that is, when an individual has the same pair of defective recessive alleles. Hence a person inherits a recessive autosomal disease only if he or she receives the defective allele from both the parents during reproduction. Examples of such disorders would include thalassemia, sickle cell anaemia, etc.
Haemophilia
Many components in the blood contribute to the formation of clots that stop bleeding. The coagulation of blood occurs with the help of a cascade mechanism and requires a number of clotting factors and calcium ions to eventually cause the conversion of soluble fibrinogen in the blood into insoluble fibrin that forms the clot and plugs the site of injury. Haemophilia is caused due to mutation in the gene coding for one of the clotting factors. Due to the absence of one of these clotting factors in the cascade pathway, the coagulation of blood does not occur and as a result, a small cut in an affected person will result in nonstop bleeding. Thus, haemophilia is also known as the bleeder’s disease.
Fig: Cascade of blood clotting
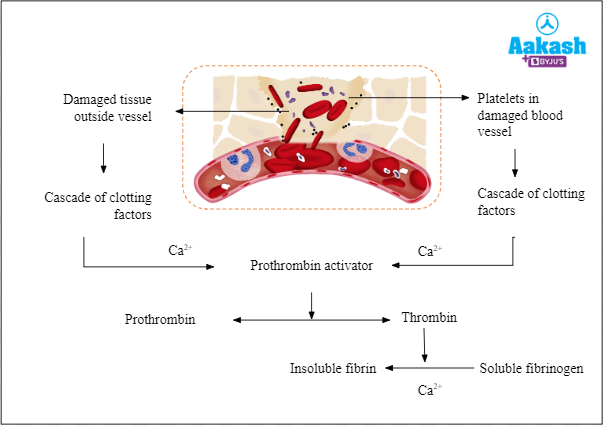
Haemophilia is classified into three types based on the factor which is affected:
- Haemophilia A: Type A haemophilia is caused by a deficiency of the blood clotting factor VIII. It affects about 85 percent of haemophiliacs.
- Haemophilia B: It occurs due to deficiency of factor IX.
Haemophilia is a sex-linked disease and it occurs due to the absence of anti-haemophiliac globulin or factor VIII (haemophilia-A) or plasma thromboplastin or factor IX (haemophilia-B) which are essential for blood coagulation.
It occurs due to the presence of defective recessive sex-linked gene ‘h’ which is carried by X-chromosome. If both X-chromosomes carry the ‘h’ gene, then the female becomes haemophilic (XhXh). The combination of these two recessive alleles is deadly and such females usually die before birth. If a female has only one allele for haemophilia, they are known as carriers and appear normal (XXh). Because the Y-chromosome lacks any corresponding allele, the presence of a single defective ‘h’ gene in males can cause the disease (XhY). Such a person requires regular blood transfusion or the required clotting factors to lead a normal life.
Now let us consider three scenarios to help us understand the inheritance of this disease better.
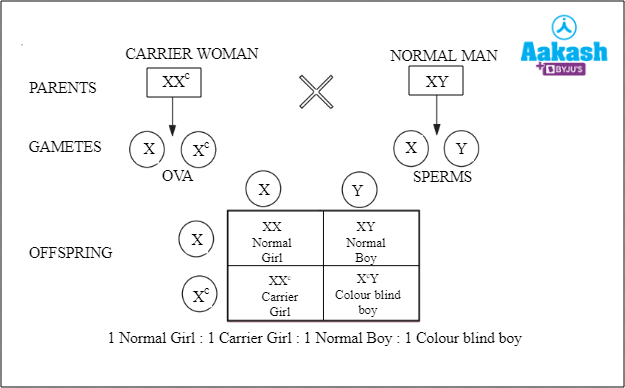
- Case I: When a carrier woman (XXh) mates with a normal man (XY)
Fig: Carrier woman is crossed with a normal man
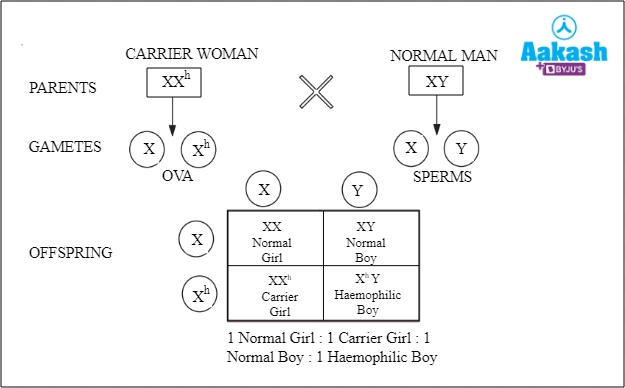
The offspring obtained in this condition can either be a normal girl, a normal boy, a carrier girl or a haemophilic boy, each having 25% probability of occurrence.
- Case II: When a normal female (XX) mates with a haemophilic male (XhY).
Fig: Normal female is crossed with a haemophilic male

The offspring obtained can either be normal males or carrier females, each having 50% chances of occurrence.
- Case III: When a carrier woman (XXh) mates with a haemophilic man (XhY).
Fig: Carrier woman is crossed with a haemophilic man
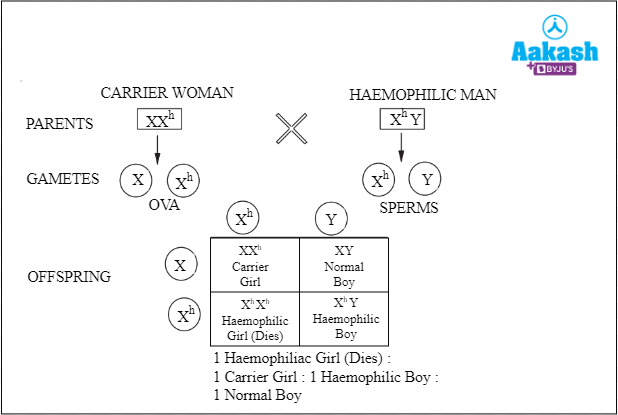
The offspring obtained in this condition can be a haemophilic girl (25% chance) who will die before birth, one carrier girl (25%), one haemophilic boy (25%) or one normal boy (25%).
Haemophilia is also referred to as Royal disease because it is common in royal families of Europe. The disease spread in the royal families through the children of Queen Victoria. The queen's ancestors were not infected with the disease. The haemophilia gene appears to have originated through mutation in either her father's or her own germ cells. Haemophilia is a sex-linked disease and due to this, it shows criss-cross inheritance because the defective gene of the mother is always inherited by the sons and the defective gene in the father is always inherited by the daughters. Males are more likely to develop haemophilia than females because in males even the presence of a single defective gene can cause the disease, whereas in females two copies of the defective gene is required for the disease to be expressed.
The Christmas disease
Christmas disease is another name for haemophilia B. This is because this disorder was first reported in a five year old patient in 1952. Stephen Christmas was the patient's name.
Fig: Stephen Christmas

Colour blindness
Colour blondness is a recessive sex-linked trait. The colour blind person is unable to distinguish red and green colours. The vision of a person is not affected. The gene responsible for normal vision is generally dominant. The normal gene and the recessive allele, both are carried by X-chromosomes. A female become colour blind, when both the sex chromosomes carry recessive gene (XcXc). As the disease is not lethal, colourblind females survive and do not die before birth.
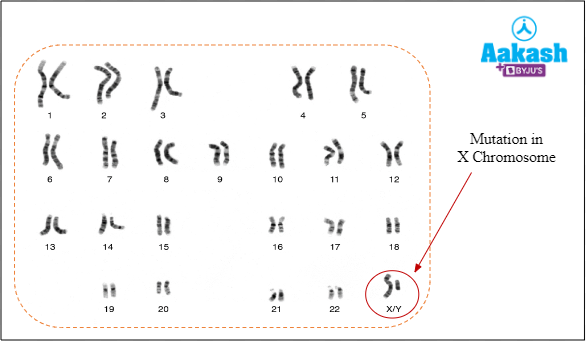
It is a sex-linked recessive disorder that occur due to defect in either red or green cone cells of an eye. As a result, a person is unable to distinguish between red and green colour. It results from a mutation in a gene, responsible for colour vision, in the X-chromosome.
Fig: Mutation in X-chromosome
There are two types of cells in the human eye that help in vision. These are rod cells and cone cells. The rod cells help in vision in dim and dark conditions and the cone cells help in colour vision under bright light. Cone cells are able to differentiate between red, blue and green colour. Colour blindness occurs when any of these cone cells fail to perform their function.
Fig: Rods and cones

The females are usually carriers of colour blindness. Because males have only one X-chromosome and females have two, males are more likely to develop colour blindness. In males even the presence of a single defective gene can cause the disease, whereas in females two copies of the defective gene are required for the disease to be expressed.
Let us consider some cases to understand the inheritance of colour blindness.
- Case I: When a carrier mother (XXc) mates with an unaffected father (XY). The offspring obtained can be either a carrier daughter, a colour blind son, a normal daughter or a normal son. The son of a carrier woman has a 50% chance of being colour blind. However, for a daughter to be colour blind, both parents should carry the colour blind gene.
Fig: Carrier mother crossed with unaffected father
- Case II: When a carrier mother (XXc) mates with the affected father (XcY), the offspring obtained are either an affected daughter, an affected son, a carrier daughter or an unaffected son. The sons have 50% chance of being colourblind or normal and the daughters have 50% chance of either being colour blind or a carrier.
Fig: Carrier mother is crossed with affected father

Sickle cell anaemia
Sickle cell anaemia is an autosomal hereditary disorder which shows incomplete dominance at the RBC level and codominance at the haemoglobin level. Haemoglobin is composed of two α chains and two β chains. Sickle cell anaemia occurs due to a mutation in the gene that codes for the β chain of the Haemoglobin (Hb) protein and is present on the 11th pair of autosomes in humans. The normal gene is notified as HbA and the mutant defective gene is notified as HbS.
In sickle cell anaemia RBCs become sickle-shaped due to a single change in amino acid in the β chain of the Haemoglobin (Hb) protein. The changes occur at the sixth position of the beta globin chain. In normal haemoglobin, glutamic acid is present which is replaced by valine in sickle cell anaemia. The mutant Haemoglobin-S protein acts like the normal Haemoglobin-A protein except under conditions of oxygen deficiency such as during exercise or high altitude. Oxygen stress creates hydrophobic bonds between the valine of the β chains and the other chains of the Hb-S protein. This distorts the haemoglobin structure and causes the erythrocytes (RBCs) to lose their spherical biconcave shape and become sickle shaped.
Fig: Cell shape in sickle cell anaemia

Sickle shaped erythrocytes lose membrane flexibility and are incapable of passing through blood capillaries and end up clogging them. This reduces blood supply to vital organs. The sickle shaped erythrocytes are also destroyed in some time, causing a deficiency of erythrocytes in the body which reflects in the form of anaemia. Jaundice, muscle cramps, headache, damage to spleen, brain and other vital organs, etc are some of the other side effects of having Hb-S protein.
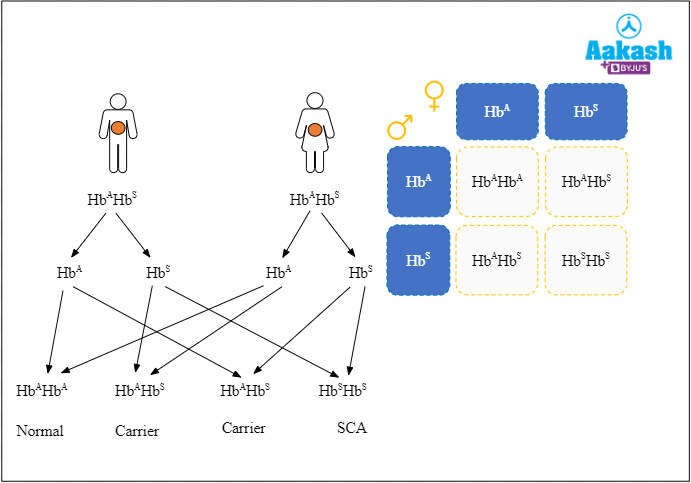
HbA and HbS genes are codominant and in heterozygous condition (HbAHbS), both express themselves. Such individuals are considered as carriers and live normally, except under conditions of oxygen deficiency. A person becomes sickle cell anaemic if they inherit the abnormal genes from both the parents and have a homozygous genotype (HbSHbS). The erythrocytes in such persons become sickle shaped even without oxygen stress and the individuals generally die before attaining maturity.
Fig: Genotype of sickle cell anaemia

Let us consider three cases of how sickle cell anaemia is inherited:
- Case I: When a carrier individual is crossed with another carrier individual, out of the four possible phenotypes in the offspring, two are carriers, one is normal and the other one is sickle cell anaemic. Thus, there is a 50% chance that the individuals will be carriers and 25% chance of being normal or affected. The suffering from the death of a severely anaemic affected child can be avoided by discouraging marriages between heterozygous carrier individuals. Heterozygous individuals can be easily recognised by blood smears.
Fig: Carrier male is crossed with carrier female
- Case II: When an affected person mates with a carrier individual, the offspring obtained are either carriers or affected, having a 50-50 chance for each. However, this case rarely occurs as individuals affected with sickle cell anaemia rarely live long enough to mate and have a family.
Fig: Affected male is crossed with carrier female

- Case III: When an unaffected person mates with the carrier female, the offspring can either be normal or carriers.
Fig: Unaffected male is crossed with carrier female

In human communities that are exposed to malaria, the sickle cell allele is present at a reasonably high frequency as it has a selective advantage in the process of natural selection in such communities. Plasmodium, the protozoan genus that causes malaria, spends part of its life cycle in the Anopheles mosquito and part of it in the RBCs of humans infected by an infected mosquito. It is however, unable to thrive in sickle shaped RBCs. Thus, heterozygous carriers of the HbS gene are benefitted and become less susceptible to malaria infection.
Thalassemia
Cooley discovered thalassemia, but Whipple and Bradford coined the phrase after it became widely known in the Mediterranean. It is an autosomal recessive blood disease that appears in those offsprings whose parents are carriers or heterozygous parents. The defect can be caused by a mutation or deletion of the genes that govern the synthesis of haemoglobin globin chains, alpha and beta chains. Anaemia is caused by an unbalanced production of haemoglobin’s globin chains.
Haemoglobin is an essential molecule that is responsible for transporting oxygen and carbon dioxide throughout the body. Structurally, Hb is composed of four polypeptide chains. Two chains are α globin chains and the other two are β globin chains.
Fig: Haemoglobin

Thalassemia induces anaemia by causing the excessive breakdown of red blood cells that in turn develops due to imbalance synthesis of globin chains. The excess globin chains remain free, insoluble, forming precipitate which damages the red blood cells and causes their lysis. Thalassemia is further divided into three types based on the type of polypeptide chain affected:
- Alpha thalassemia - decreased synthesis of α globin
- Beta thalassemia - decreased synthesis of β globin
- Delta thalassemia - This defect is of minor nature because delta globin occurs in only 3% of adult haemoglobin as alpha and delta tetramers. The gene controlling the synthesis of delta globin is the HBD gene situated on chromosome 11.
Alpha thalassemia
As the name suggests, alpha thalassemia occurs when there is a defective formation of alpha globin chain. In this disorder, there is decreased synthesis of α globin, resulting in an excess of β globin chains in adults and 𝛾 (gamma) chains in newborns. As a result, haemoglobin tetramers are unstable and there is abnormal oxygen dissociation curve.
This condition is controlled by two genes that are present on chromosome 16. These two genes are HBA1 and HBA2 and they collectively have four alleles. Silent carriers have one defective allele, while those with two defective alleles have thalassemia minor with mild anaemia.
Fig: Alpha thalassemia

A person with three defective alleles is likely to develop the haemoglobin H or HbH disease which is characterised by anaemia, enlargement of spleen and liver, jaundice, overgrowth of upper jaw and forehead, etc.
In case, all the four alleles are defective, the foetus develops hydrops foetalis which is an excessive buildup of fluid in the foetal body. This is accompanied with anaemia, enlarged liver and spleen, heart defects, abnormalities in the urinary system and genitalia. The mother often develops high blood pressure with swelling. Delivery is generally premature and associated with abnormal blood loss. The child is either still born or dies soon after delivery.
Beta thalassemia
In this condition, there is a decreased synthesis of β globin chain. The defect occurs on the alleles of the HBB gene present on chromosome 11. Thalassemia minor is characterised by a higher number of microcytic (smaller than usual) erythrocytes and a lower amount of haemoglobin in those who have one faulty allele. On the contrary, if a person has both the defective alleles then it results in Cooley’s anaemia or thalassemia major.
Fig: Beta thalassemia

Due to excess of α globin chains, the Hb tetramer formation is prevented. The excess globins tend to bind to RBC membranes, causing their damage and lysis which leads to anaemia. At high concentration, toxic aggregates are formed. The disease manifests itself after 4-6 months of birth. There is severe haemolytic microcytic hypochromic (less red than usual RBCs) anaemia along with enlargement of spleen and liver (hepatosplenomegaly), skeletal deformation, cardiac enlargement and mongoloid facial features.
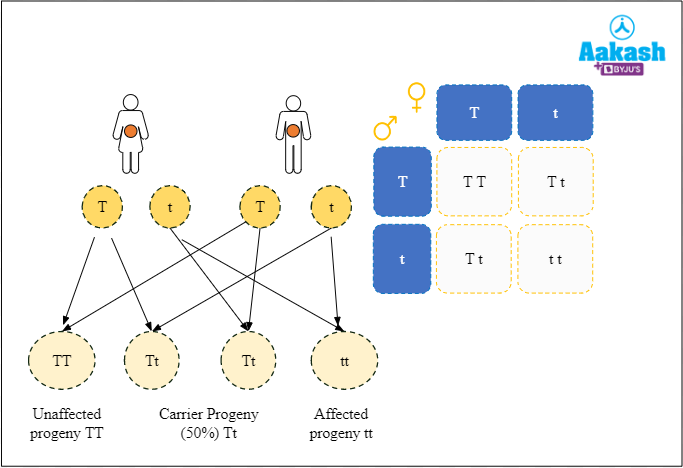
Let us consider three cases to understand the inheritance of thalassemia.
- Case I: When a carrier individual mates with another carrier individual, there is 50% chance that the offspring will be a carrier, 25% chance that he or she will be unaffected and 25% chance that the offspring will be affected with thalassemia. Inheritance of thalassemia from carrier parents is the most common form of inheriting thalassemia and can be prevented by discouraging carrier individuals from mating. Carriers can be identified using simple blood tests.
Fig: Carrier female is crossed with a carrier male
- Case II: When a carrier individual mates with an unaffected individual, there are 50% chances of the offspring either being carriers or being completely unaffected.
Fig: Carrier female is crossed with unaffected male

- Case III: When an affected individual mates with a carrier individual, the offspring progeny has 50% chances of either being affected or being carrier. This rarely happens because individuals affected with thalassemia major rarely live up to mating age.
Fig: Affected female is crossed with carrier male

- Case IV: When an unaffected individual mates with an affected individual, all the offspring produced are carriers.
Fig: Affected female is crossed with an unaffected male

Phenylketonuria
It is a metabolic disorder that occurs due to lack of an enzyme phenylalanine hydroxylase (PHL). It is an autosomal recessive disorder. The enzyme phenylalanine hydroxylase is essential for the conversion of phenylalanine to tyrosine. Tyrosine is a necessary amino acid for the production of a variety of neurotransmitters. Reduced brain development and other neurological problems are the outcome of deficiency of tyrosine.
Fig: Role of PHL

In the absence of phenylalanine hydroxylase enzymes, the concentration of phenylalanine increases. The accumulation of phenylalanine becomes toxic.
Symptoms
The symptoms of phenylketonuria are listed below:
- Learning disability and delayed development
- Neurological disorders
- Small-sized head and this condition is known as microcephaly
- Neurological disorders
- Behavioural, emotional and social problems
Fig: Microcephaly

Let us consider three cases to understand the inheritance of phenylketonuria.
- Case I: When a carrier individual mates with another carrier individual, the offspring has 50% chance of being a carrier, 25% chance of being unaffected and 25% chance of being affected.
Fig: Carrier female is crossed with a carrier male

- Case II: When an unaffected individual mates with a carrier individual, the offspring's progeny has 50% chances of either being unaffected or being carrier.
Fig: Unaffected female is crossed with a carrier male

- Case III: When an unaffected individual mates with an affected individual, all the offspring produced are carriers.
Fig: Unaffected female is crossed with an affected male

Cystic fibrosis
It is due to an autosomal recessive gene present on chromosome 7 that controls the synthesis of a unique glycoprotein. It results in the production of very high viscosity mucus and excessive secretion of sodium in sweat. The disease affects many exocrine glands such as glands of the skin, lungs, liver, pancreas. Excessive mucus in lungs causes breathing difficulties and makes the lung more susceptible to infections. Bile and pancreatic juice production is impaired, the liver may undergo cirrhosis and all of this collectively affects digestion. Cardiac failure is also common.
Practice Problems
1. Mendelian disorders occur due to:
- abnormal shape of a single chromosome
- mutation in the single gene
- presence of an extra chromosome
- absence of a particular chromosome
Solution: Mendelian disorders occur mainly by the alteration or mutation in a single gene. Pedigree analysis can be used to trace the pattern of inheritance of Mendelian disorders in a family. Some examples of Mendelian disorders are haemophilia, sickle cell anaemia and colour blindness. Hence, the correct option is b.
2. Identify the sex-linked recessive trait
- Sickle - cell anaemia
- Haemophilia
- Cystic fibrosis
- Both a and b
Solution: Haemophilia is a sex-linked recessive disease which is also termed as bleeder’s disease. A haemophiliac person will continue to bleed even after a minor cut. This occurs due to the absence of anti-haemophiliac globulin or factor VIII (haemophilia-A) or plasma thromboplastin or factor IX (haemophilia-B) which are essential for blood coagulation. The absence of either of these clotting factors is caused due to a mutation in the gene responsible for their production. As this gene is present on the X chromosome, the disorder is sex-linked in nature. The mutant gene is recessive to the normal gene. Hence, the correct option is b.
3. If a colour blind man marries a non-colour blind daughter of a colour blind father, the offspring obtained will be
- None of their daughters will be colour blind
- All the sons will be colour blind
- All the daughters will be colour blind
- Approximately half of their sons will be colorblind.
Solution: Colour blondness is a recessive sex-linked trait. The colour blind person is unable to distinguish red and green colours. The vision of a person is not affected. The gene responsible for normal vision is generally dominant. The normal gene and the recessive allele, both are carried by X-chromosomes. In the given condition, the genotype of colour blind man is XcY. As colour blindness is a X linked disorder, it’s inheritance is criss cross in nature, that is, the defective gene of the father will be inherited by the daughter. As the female partner is born to a colourblind father but is herself not colour blind, she must be a carrier having received the defective allele from the father and the normal allele from the mother. Thus, her genotype will be XcX. When both are crossed, the offspring obtained are:
Fig: Cross between colour blind man and carrier woman

This shows that half of their sons will be colour blind. Hence, the correct option is d.
4. A haemophilic man marries a woman who is heterozygous for the disease. In the F1 generation, what will be the ratios between carrier daughters, haemophilic daughters, normal sons, and haemophilic sons?
- 1: 2: 2:1
- 2: 1: 1: 2
- 1: 1: 1: 1
- 1: 2: 1: 2
Solution: Haemophilia is a sex-linked disease and is also termed as bleeder’s disease. The haemophiliac person will continue to bleed even after a minor cut. This occurs due to the absence of anti-haemophiliac globulin or factor VIII (haemophilia-A) or plasma thromboplastin or factor IX (haemophilia-B) which are essential for blood coagulation. It occurs due to the presence of recessive sex-linked gene ‘h’ which is carried by X-chromosome. In the given condition, the genotype of heterozygous female is XXh and the genotype of haemophilic male is XhY. The cross between these two produced offsprings are as follows:
Fig: Carrier mother is crossed with affected father

From the above cross, the ratio obtained is 1:1:1:1. Hence, the correct option is c.
5. What is the distinction between sickle cell anaemia and thalassemia?
Solution: Thalassemia is a quantitative problem in which there is an inadequate synthesis of globin molecules. On the other hand, sickle cell anaemia is a qualitative problem in which a single amino acid change in the β globin chain of haemoglobin distorts its structure and results in formation of sickle shaped RBCs.
6. What are the symptoms of haemophilia?
Solution: Haemophilia is a sex-linked disease which is also termed as bleeder’s disease. The haemophiliac person will continue to bleed even after a minor cut. This occurs due to the absence of anti-haemophiliac globulin or factor VIII (haemophilia-A) or plasma thromboplastin or factor IX (haemophilia-B) which are essential for blood coagulation. The symptoms of haemophilia are as follows:
- Excessive bleeding from the injury site
- Deep bruises
- Unusual bleeding
- Blood in urine or stool
7. Why is haemophilia a rare condition in females?
Solution: Haemophilia is a rare condition in females because of its inheritance pattern. It is a sex-linked recessive disease. It occurs due to the presence of recessive sex-linked gene ‘h’ which is carried by X-chromosome. If both X-chromosomes carry the ‘h’ gene, then the female becomes haemophilic (XhXh). Because the combination of these two recessive alleles is deadly, such females usually die before giving birth. If a female has only one allele for haemophilia, they are known as carriers and appear normal (XXh). Because the Y-chromosome lacks any corresponding allele, a single gene causing the haemophilia can express itself in males (XhY).
FAQs
1. What is tritanopia?
Answer: Tritanopia is blue colour blindness in which the individual is unable to distinguish between blue and yellow colours. It is rare and is generally caused by drug toxicity, retinal detachment and disorders of the nervous system.
2. What is Daltonism?
Answer: Red-green colour blindness is also known as Daltonism because the famous scientist Dalton also suffered from this disorder.
3. What is the total number of Mendelian diseases?
Answer: There are about 7,300 of Mendelian disorders, which are rare and usually inherited and are caused by a single gene mutation.
4. Are the majority of human characteristics inherited by Mendelian patterns?
Answer: Most human features are governed by many genes or contain more than two alleles, indicating that they are not inherited in a simple Mendelian pattern.
5. What is HPLC?
Answer: HPLC or High Performance Liquid Chromatography is a technique used to separate, identify and quantify components of a mixture. Pumps are used to pass a liquid solvent containing the mixture, under pressure, through a column which is filled with a solid adsorbent material. The components of the mixture have unique interactions with the adsorbent material, which causes each of them to flow through the adsorbent column at different rates and causing them to separate out. This technique is widely used in the screening of thalassemia carriers by separating out and quantifying the normal and defective haemoglobin proteins in the blood.
Related Topics:
|
Chromosomal theory of inheritance: Sutton and Boveri Experiments, Practice Problems and FAQs |
|
Law of Segregation: Dihybrid cross, Practice Problems and FAQs |
|
Law of Dominance Law of Dominance, Practice Problems and FAQs |
|
Incomplete dominance ,Codominance,practice problems and FAQs |
|
Multiple Allelism,Practice problems and FAQs |
|
Sex determination: Genotypic and environmental sex determination, Genic balance theory of sex determination, Practice Problems and FAQ’s |